
 Prions are not viruses – they are infectious proteins that lack nucleic acids. Nevertheless, virologists have always been fascinated by prions – they appear in virology textbooks (where else would you put them?) and are taught in virology classes. I’ve written about prions on this blog (five articles, to be exact – look under P in the Table of Contents) and I’m fascinated by their biology and transmission. That’s why the newly solved structure of an infectious prion protein is the topic of the sixth prion article at virology blog.
Prions are not viruses – they are infectious proteins that lack nucleic acids. Nevertheless, virologists have always been fascinated by prions – they appear in virology textbooks (where else would you put them?) and are taught in virology classes. I’ve written about prions on this blog (five articles, to be exact – look under P in the Table of Contents) and I’m fascinated by their biology and transmission. That’s why the newly solved structure of an infectious prion protein is the topic of the sixth prion article at virology blog.
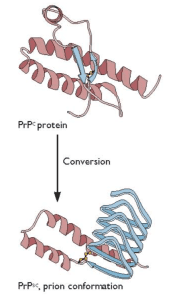
Spongiform encephalopathies are neurodegenerative diseases caused by misfolding of normal cellular prion proteins. Human spongiform encephalopathies are placed into three groups: infectious, familial or genetic, and sporadic, distinguished by how the disease is acquired initially. In all cases, the pathogenic protein is the host-encoded PrPC protein with an altered conformation, called PrPsc. In the simplest case, PrPSc converts normal PrPC protein into more copies of the pathogenic form (illustrated).
The structure of the normal PrPC protein, solved some time ago, revealed that it is largely alpha-helical with little beta-strand content. The structure of PrPSc protein has been elusive, because it forms aggregates and amyloid fibrils. It has been suggested that the PrPSc protein has more beta-strand content than the normal protein, but how this property would lead to prion replication was unknown. Clearly solving the structure of prion protein was needed to fully understand the biology of this unusual pathogen.
The structure of PrPSc protein has now been solved by cryo-electron microscopy and image reconstruction (link to paper). The protein was purified from transgenic mice programmed to produce a form of PrPSc protein that is not anchored to the cell membrane, and which is also underglycosylated. The protein causes disease in mice but is more homogeneous and forms fibrillar plaques, allowing gentler purification methods.
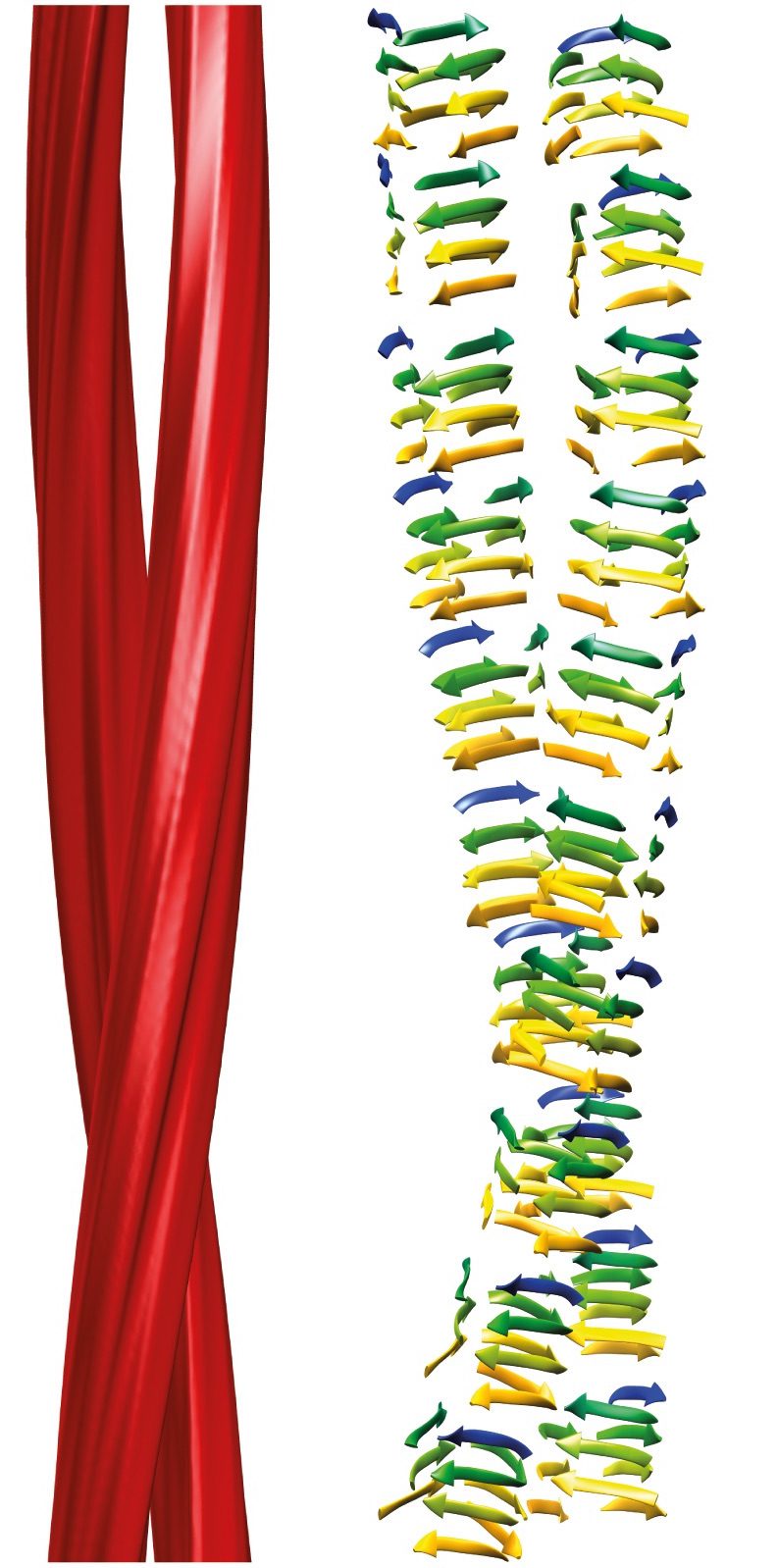 The structure of this form of the PrPSc protein reveals that it consists of two intertwined fibrils (red in the image) which most likely consist of a series of repeated beta-strands, or rungs, called a beta-solenoid. The structure provides clues about how a pathogenic prion protein converts a normal PrPC into PrPSc . The upper and lower rungs of beta-solenoids are likely the initiation points for hydrogen-bonding with new PrPC molecules – in many proteins with beta-solenoids, they are blocked to prevent propagation of beta-sheets. Once added to the fibrils, the ends would serve to recruit additional proteins, and the chain lengthens.
The structure of this form of the PrPSc protein reveals that it consists of two intertwined fibrils (red in the image) which most likely consist of a series of repeated beta-strands, or rungs, called a beta-solenoid. The structure provides clues about how a pathogenic prion protein converts a normal PrPC into PrPSc . The upper and lower rungs of beta-solenoids are likely the initiation points for hydrogen-bonding with new PrPC molecules – in many proteins with beta-solenoids, they are blocked to prevent propagation of beta-sheets. Once added to the fibrils, the ends would serve to recruit additional proteins, and the chain lengthens.
The authors note that the molecular interactions that control prion templating, including hydrogen-bonding, charge and hydrophobic interactions, aromatic stacking, and steric constraints, also play roles in DNA replication.
The structure of PrPSc protein provides a mechanism for prion replication by incorporation of additional molecules into a growing beta-solenoid. I wonder if incorporation into fibrils is the sole driving force for converting PrPCprotein into PrPSc, or if PrPC is conformationally altered before it ever encounters a growing fibril.
To know more about prions, which book or reading is recommended?