
Early in the COVID-19 pandemic, SARS-CoV-2 genome variation appeared to be low, with an average of 10 base differences in the 30,000 base genome between any two isolates. Late in 2020, as many more people were infected, variants were isolated that had more changes than previously seen. A study of variation at the genome level indicates that diversity in a single host has been underestimated.
SARS-CoV-2 variants certainly arise in each infected individual, and these may be subjected to selection pressures such as those imposed by antibodies binding to spike protein. Commonly used sequencing technologies do not permit detection of virus variants within single samples. To address this limitation, a high-throughput sequencing method was developed for sequencing individual virus RNA genomes in patient samples. The results demonstrate that SARS-CoV-2 variants emerge in each host and are subject to immune selection pressure.
When this method was used to analyze a patient isolate of SARS-CoV-2 after four passages in Vero cells, in the regions encoding spike, ORF3, envelope, and M protein, 18 unique combinations of mutations were detected. Over half of the single genome sequences differed from the reference sequence. Many of the changes led to amino acid substitutions, suggesting selection pressure occurring during cell culture adaptation.
In-host diversity was then examined for 7 patients sampled between days 8 and 17 of illness onset. Only a few sequence haplotypes were detected, with no clear consensus among the amino acid changes. These relatively homogeneous sequences might have been a consequence of sampling early in infection when limited antibody pressure was present. Examination of the antibody response in longitudinal samples revealed an increasing and broadening antibody response to the spike.
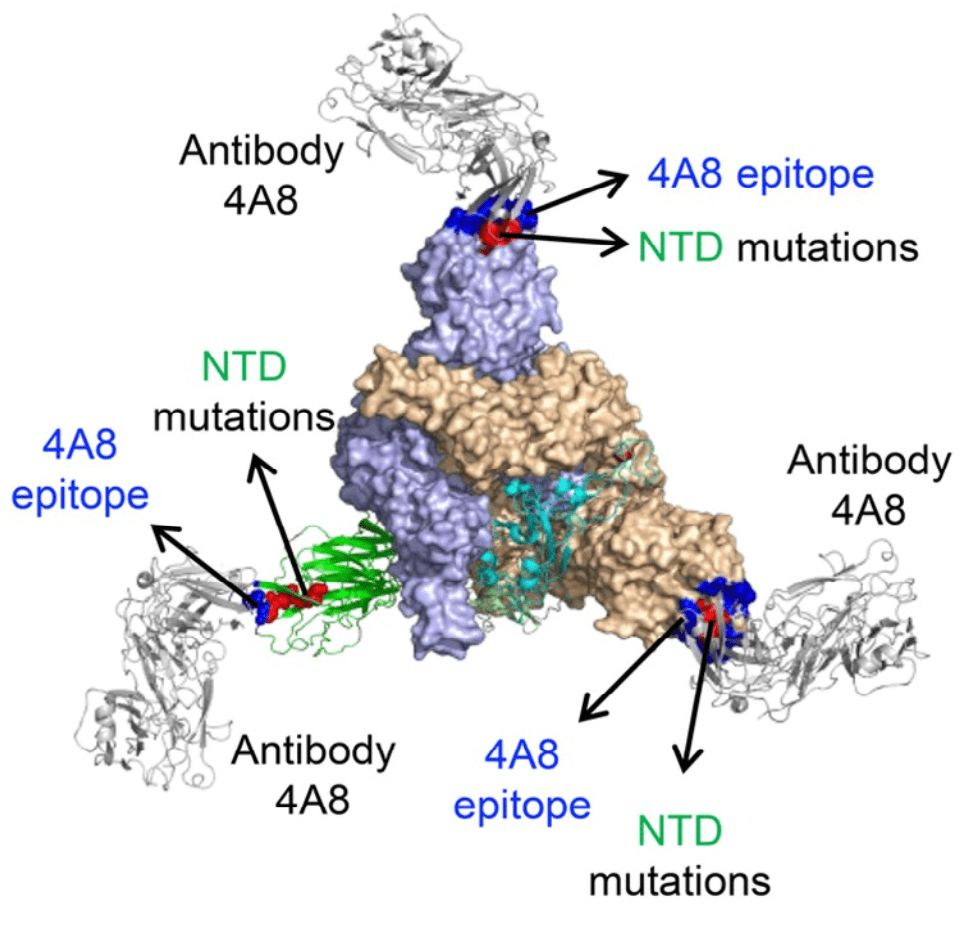
A study of antibody responses and presence of mutations in individual genomes revealed a correlation between the emergence of antibodies to the N-terminal domain of spike protein, the emergence of amino acid changes in regions that bind these antibodies, and a delay in clearance of virus. These results are consistent with antibody pressure during infection leading to selection of SARS-CoV-2 spike variants (pictured).
SARS-CoV-2 variation in a single host had not been previously described, probably due to the inability of previous sequencing methods to distinguish variation from technical errors. It is also likely that high-quality data were acquired early in infection when viral titers are high yet antibody pressure is not yet present.
There are several take-home messages from these findings. They illustrate how diversity in a single host might give rise to antibody-resistant variants that could spread in a population if they have the appropriate fitness. In-host escape might be avoided by containing viral reproduction early in infection, either by prior immunity or by antiviral therapy, before selection by antibody takes place. This method should be applied to studying more COVID-19 patient samples to understand the extent of potential in-host SARS-CoV-2 variation.
MUST READ
I was diagnosed with Fibromyalgia years ago after long treatment with various drugs (illness progress whole that time). I started on Worldherbsclinic Fibromyalgia HERBAL REMEDY called FB FORMULA two months ago. After THREE MONTHS of usage my Fibromyalgia stopped progressing and was totally reversed. WORLDHERBSCLINIC FB FORMULA is everything to me and the best for Fibromyalgia patients !!
Since virus clearance is caused by CD-8 T-cells, not Ab’s, there should be some interaction.
A fascinating radiolab podcasts suggests that variants are partly driven by medical treatments.
The UK-guy in the story had a suppressed immune system from cancer and the treatment he was receiving for it. But, as they were treating him for Covid, they kept sequencing the virus, and they watched as about a dozen variants arose and exploded in his body…until he died around day 102. He received 2 Remdesivir treatments and 2 treatments with convalescent plasma.
https://www.wnycstudios.org/podcasts/radiolab/articles/dispatch-14-covid-crystal-ball
Pingback: SARS-CoV-2 variants arise during individual infections - Virology Hub