
 by Gertrud U. Rey
by Gertrud U. Rey
A small subset of people are more vulnerable to severe complications and death resulting from SARS-CoV-2 infection. What causes this vulnerability?
An increasing body of evidence suggests that patients suffering from severe COVID-19 often have one or more pre-existing conditions (co-morbidities). In an effort to describe the co-morbidities of COVID-19 patients requiring hospitalization, a recent case series tracked the course of disease in 5,700 patients in 12 different New York City hospitals over a period of five weeks. The study revealed that the median age of the hospitalized patients was 63, and that 57% of these patients had hypertension, 34% had diabetes, and 42% were obese. Interestingly, the vast majority of the patients (88%) had two or more of these co-morbidities.
While these underlying diseases are clearly risk factors for severe COVID-19, the exact mechanisms responsible for the morbidity and mortality in the affected patients are unclear. Nonetheless, the overwhelming amount of literature continuously emerging on this topic provides some insight.
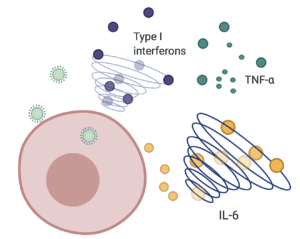
Sentinel cells of the innate immune system can sense the presence of viruses never seen before and trigger a cascade of events that mobilizes immune cells such as macrophages, neutrophils, and dendritic cells to the site of infection. Once there, these immune cells produce pro-inflammatory signaling proteins known as cytokines, which then cue other responses and prime adaptive T and B cells for future functions. A primary wave of cytokines includes type I interferons, which stimulate a signaling cascade that ultimately limits viral replication. As the infection progresses and the virus is cleared, the recruited immune cells and resulting cytokines typically recede and the patient recovers. However, in some cases the immune reaction continues, leading to an excessive inflammatory response often referred to as a “cytokine storm.”
In an effort to define some of the key players in SARS-CoV-2-associated cytokine storms, a group in France analyzed the cytokine responses in fifty COVID-19 patients experiencing varying disease severity. The authors found that patients with severe disease had persistently high levels of virus in the blood – a condition known as viremia, and a significantly impaired type I interferon response. In contrast, patients with mild to moderate disease exhibited low viremia and a high type I interferon response. In parallel, patients with severe disease had increased levels of the pro-inflammatory cytokines tumor necrosis factor-α and interleukin-6 (IL-6), both of which are major promoters of fever and other tissue changes that occur in response to cytokine production. The authors suggest that the acute respiratory distress syndrome seen in patients with severe COVID-19 may be triggered by a combined effect of these pro-inflammatory cytokines and impaired type I interferon responses.
The authors also note that critically ill patients produced lower levels of T cells, an effect that aligns with observations that SARS-CoV-2 can infect and kill T cells. Because T cells are crucial for directly killing infected host cells and regulating and/or suppressing the immune response, this finding would further explain why patients with severe disease have more inflammation.
Increased trafficking of pro-inflammatory cytokines can also lead to dysfunctions in blood clotting. Normal blood clotting typically involves the aggregation of clotting factors to limit excessive bleeding and trap microorganisms. SARS-CoV-2 infection can cause thrombosis, the formation of small clots throughout the lungs, which can restrict blood flow and thus oxygenation of the blood in the lungs. Sometimes, pieces of the clots can break off, lodge in vessels, and cause life-threatening obstruction of blood flow to the brain or other vital organs, thereby compromising their function. The digestion of clots releases small protein fragments called D-dimers, which are being used as biomarkers of severe disease and are associated with an increased risk of death in COVID-19 patients. As clots form and break down, the supply of clotting factors is depleted, leading to leaky blood vessels and movement of fluid into the lungs and other tissues.
As we gain a better understanding of SARS-CoV-2 pathogenesis, it may be necessary to track disease progression in high-risk patients by monitoring the presence of biomarkers like tumor necrosis factor-α, IL-6, and D-dimers. It is possible that co-morbidities associated with severe COVID-19 also prevent production of type I interferon and exacerbate inflammation. For example, obesity can lead to reduced type I interferon responses. The severity of disease in patients with comorbidities could be mitigated with administration of interferon and/or anti-inflammatory therapies that target IL-6 or tumor necrosis factor-α. Clinical trials are currently underway to evaluate the benefits of IL-6 targeted therapy in the context of COVID-19. Also, if D-dimers are detected in a timely manner, patients can be treated with heparin or other blood thinners to prevent thrombosis, which might improve COVID-19 outcomes. Further studies are needed to identify additional drugs that can be used to target key inflammatory markers in SARS-CoV-2 infection.
All things considered, only about 95% of severe COVID-19 cases are accounted for by old age or the co-morbidities described above. There is a small subset of individuals under the age of 50 who become very ill and die from SARS-CoV-2 infection even in the absence of obvious co-morbidities. Experts believe that certain genetic factors may predispose younger, previously healthy individuals to severe disease. For a review of this topic, I recommend this interview with Jean-Laurent Casanova of Rockefeller University.
[Cindy Leifer provides an excellent explanation of blood clotting in the context of SARS-CoV-2 infection on episode 30 of Immune.]
Pingback: The perfect storm – Virology Hub
Bahag
There is always a clear mention of hypertension as a very significant risk factor in severe disease with SARS-CoV-2. Does this assume the hypertension is well controlled by drug therapy or are they referring to untreated hypertension? Does successful control of hypertension make any positive difference to severity of disease ?
40% of Americans over age 20 are obese. Source: https://www.cdc.gov/nchs/fastats/obesity-overweight.htm
45% of American adults have hypertension. Source: https://www.cdc.gov/bloodpressure/facts.htm For the 55+ age group I’d bet money, without Googling, it’s ~57%.
Those seem less co-morbidities than a reflection of the population. (That is, consequential to death rates in America as against the world, but not necessarily a distinguisher for SARS-CoV-2 lethality.)
The diabetes prevalence seems maybe ~2x higher than would be expected, however, unless the 34% figure includes pre-diabetes and borderline cases. For age distributions see https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3464829/
It was pointed out to me that I was comparing the general American population to the New York City cohort. New York City has a lower prevalence of obesity, etc. Though for the age groups that gap smaller than for all adults. For example, prevalence of obesity in the NYC 65-74 age group is 31%. See https://www1.nyc.gov/assets/doh/downloads/pdf/episrv/2019-older-adult-health.pdf page 23. And if you adjust for race, poverty, etc, the gap closes.
Even if we assume that the disproportionate impact of the virus on certain racial and economic groups is completely unrelated to mitigation compliance (and thus the higher rate of deaths in such groups wholly contributable to these physiological co-morbidities), I’m still not sure what’s to be gleaned by focusing on them, particularly in search of peculiar viral pathogenesis. If the cohort were from the U.S. at large they would be unremarkable. Obesity, etc, are co-morbidities for almost all causes of death. And while it’s useful to understand how the effect disease progression, it’s not prima facie useful for understanding how COVID-19, specifically and uniquely, progresses.
Do we know if SARS-CoV-2 infects T cells? If so is it abortive? Or are there simply reduced T cell counts in the severe patients