
By Gertrud U. Rey
Zika virus (ZIKV) infection causes microcephaly in newborns and is causally associated with Guillian-Barré syndrome in adults. To date, there are no drugs available to prevent or treat ZIKV infection. ZIKV vaccine research is challenging because adult immunocompetent mice are resistant to ZIKV infection and disease.
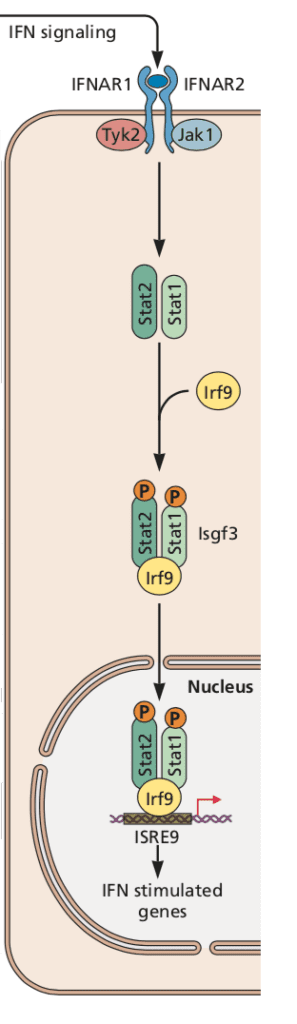
The primary immunologic response to ZIKV infection is the production of type I interferon, which binds to the interferon alpha/beta 1 receptor (IFNAR1) and activates signal transducer and activator of transcription (STAT) complexes (illustrated). Activation of this pathway leads to the production of hundreds of antiviral and immunomodulatory interferon-stimulated genes and a general antiviral state. In humans, ZIKV evades the innate immune response by degrading STAT2, thereby antagonizing type I interferon (click here for a review of how viruses interfere with interferon). However, in mice ZIKV is incapable of binding STAT2 and triggering its downstream transcriptional effects, possibly explaining the resistance of immunocompetent mice to ZIKV. Consequently, most mouse studies of ZIKV pathogenesis have been done in mice with genetic or acquired deficiencies of interferon signaling. Unfortunately, such models don’t mimic human disease sufficiently to make them suitable for evaluating vaccines and therapeutics.
Using a two-step approach, a group at Washington University School of Medicine has developed a new system to study ZIKV infection in mice (link to paper). First, they generated a mouse-adapted ZIKV strain by passaging the African strain ZIKV-Dak (parental strain) in mice that have an intact type I interferon response but lack mature B and T cell responses. Second, they developed a fully immunocompetent mouse model of ZIKV infection by introducing human STAT2 into the mouse Stat2 locus.
After passaging the parental virus strain four times, the authors obtained ZIKV-Dak-MA (MA indicates mouse-adapted), a strain containing three mutations. The first was a silent mutation in the nucleotide sequence coding for E protein, while the other two were non-synonymous mutations of lysine to arginine, and glycine to arginine at positions 399 and 18 of the NS3 and NS4B proteins, respectively. To test the infectivity of this mutated viral strain, wild type mice were first treated with an anti-Ifnar1 monoclonal antibody and then infected. Compared to the parental strain, ZIKV-Dak-MA was more lethal, with a higher level of replication in the mouse brains.
This higher infectivity in brains may be due to an increased ability of the mutant virus to cross the blood-brain barrier or because it can replicate better in neurons. To test these possibilities, the authors infected mice intracranially with mutant or parental virus in the absence of anti-Ifnar1 antibody treatment. Intracranial infection would bypass the blood-brain barrier, and omitting anti-Ifnar1 antibody would lead to cessation of viral replication before systemic dissemination. Although neither virus caused a lethal infection, mutant viral RNA accumulated to greater levels in multiple regions of the brain, compared to parental virus RNA. In contrast, intracranial infection of mice deficient in IFNAR1 receptor with either strain resulted in minimal differences in viral RNA produced by the two strains. This result suggests that the difference in replication between the two viruses is likely mostly due to different abilities to evade type I interferon responses.
To determine which of the two non-synonymous mutations was responsible for enhanced infection and disease, the authors engineered viruses with the non-synonymous mutations in either NS3, NS4B, or both. Mice were then treated with anti-Ifnar1 monoclonal antibody and challenged with either of the mutant viruses. Viruses with mutations in either NS4B or both NS3 and NS4B were about 95% lethal in mice, while the virus with the NS3 mutation only caused about 5% mortality. The authors obtained the same result in mouse and human neural stem cells, suggesting that the NS4B mutation is necessary and sufficient for enhanced virulence.
Single-cell RNA sequencing analysis of neural stem cells infected with the parental or the NS4B mutant virus revealed that the NS4B mutant is less effective at inducing interferon-stimulated genes and type I interferon responses compared to the parental virus. To determine whether ZIKV modulates type I interferon signaling responses, the authors pre-treated murine neural stem cells with either anti-Ifnar1 antibody or exogenous IFN-b and then infected the cells with parental or NS4B mutant virus. Viral yields produced by the NS4B mutant were higher in the absence, and lower in the presence of anti-Ifnar1 antibody. However, pre-treatment of cells with IFN-b caused the two viruses to replicate to similar levels, suggesting that the NS4B mutation likely results in diminished levels of IFN-b. The fact that the NS4B mutant was still sensitive to exogenous interferon suggests that it may be unable to bind to and degrade mouse STAT2.
In the second major part of this study, the authors generated a fully immunocompetent mouse model of ZIKV infection by introducing human STAT2 into the mouse Stat2 locus. Infection of these human STAT2 knock-in mice with ZIKV-Dak-MA led to 30% mortality, with higher viral levels observed in the spleen and brain. In contrast, none of the mice inoculated with parental virus died. In addition, pregnant ZIKV-Dak-MA recipients passed the virus to their babies. Higher levels of ZIKV-Dak-MA were detected in the placenta and fetal heads of STAT2 knock-in mice. The authors concluded that combining the adapted ZIKV strain with human STAT2 knock-in mice produces a model system that mimics the human mechanism of ZIKV pathogenesis.
The lack of a suitable immunocompetent mouse model for ZIKV infection has been an impediment to research progress in this field. The model system presented here has drawbacks, such as the need to use a specific viral mutant, and a disparity of phenotype observed in peripheral organs and the brain following subcutaneous inoculation with either mutant and parental strains. However, it presents an additional resource for determining mechanisms of pathogenesis, defining correlates of innate and adaptive immune protection, and for developing drugs.
Pingback: A Mouse Model System for Zika Virus Infection - Virology