
 The high mutation rate of RNA viruses enables them to evolve in the face of different selection pressures, such as entering a new host or countering host defenses. It has always been thought that the sources of such mutations are the enzymes that copy viral RNA genomes: they make random errors which they cannot correct. Now it appears that a cell enzyme makes an even greater contribution the mutation rate of an RNA virus.
The high mutation rate of RNA viruses enables them to evolve in the face of different selection pressures, such as entering a new host or countering host defenses. It has always been thought that the sources of such mutations are the enzymes that copy viral RNA genomes: they make random errors which they cannot correct. Now it appears that a cell enzyme makes an even greater contribution the mutation rate of an RNA virus.
Deep sequencing was used to determine the mutation rate of HIV-1 in the blood of AIDS patients by searching for premature stop codons in open reading frames of viral RNA. Because stop codons terminate protein synthesis, they do not allow production of infectious viruses. Therefore they can be used to calculate the mutation rate in the absence of selection. The mutation rate calculated in this way, 0.000093 mutations per base per cell, was slightly higher than previously calculated from studies in cell culture.
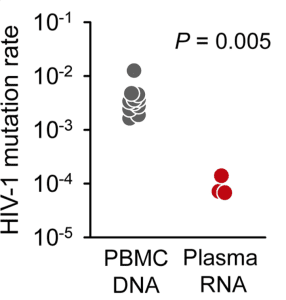
When HIV-1 infects a cell, the enzyme reverse transcriptase converts its RNA genome to DNA, which then integrates into the host cell genome. Identification of stop codons in integrated viral DNA should provide an even better estimate of the mutation rate of reverse transcriptase, because mutations that block the production of infectious virus have not yet been removed by selection. The mutation rate calculated by this approach was 0.0041 mutations per base per cell, or one mutation every 250 bases. This mutation rate is 44 times higher than the value calculated from viral RNA in patient plasma (illustrated).
Sequencing of integrated viral DNA from many patients revealed that the vast majority of mutations leading to insertion of stop codons – 98% – were the consequence of editing by the cellular enzyme APOBEC3G. This enzyme is a deaminase that changes dC to dU in the first strand of viral DNA synthesized by reverse transcriptase. APOBEC3G constitutes an intrinsic defense against HIV-1 infection, because extensive mutation of the viral DNA reduces viral infectivity. Indeed, most integrated HIV proviruses are not infectious as a consequence of APOBEC3G-induced mutations. That infection proceeds at all is due to incorporation of the viral protein vif in the virus particles. Vif binds APOBEC3G, leading to its degradation in cells.
The mutation rate of integrated HIV-1 DNA calculated by this method is much higher than that of other RNA viruses. This high mutation rate is driven by the cellular enzyme, APOBEC3G. At least half of the mutations observed in plasma viral RNAs are also contributed by this enzyme.
It has always been thought that error-prone viral RNA polymerases are largely responsible for the high mutation rates of RNA viruses. The results of this study add a new driver of viral variation, a cellular enzyme. APOBEC enzymes are known to introduce mutations in the genomes of other viruses, including hepatitis B virus, papillomaviruses, and herpesviruses. Furthermore, the cellular adenosine deaminase enzyme can edit the genomes of RNA viruses such as measles virus, parainfluenza virus, and respiratory syncytial virus. Cellular enzymes may therefore play a much greater role in the generation of viral diversity than previously imagined.
Is it actually a good defensive strategy against viral infection? I mean, if the cell promotes the viral genome mutation, very probably, there will appear resistant clones to this cellular enzyme, i guess…(sorry for my bad english). Cool article !
Like many other innate immune defenses, APOBECs target part of the pathogen’s lifecycle that is essential and highly conserved, and thus it is difficult (but not impossible) for resistant mutants to emerge; in this case, it specifically targets the DNA-RNA complexes that inevitably form when reverse transcription takes place. Since reverse transcription is an essential part of the retroviral lifecycle, HIV can’t stop doing it. There’s no specific APOBEC binding site that can be mutated, APOBEC just likes to bind RNA in general. Instead, HIV has had to develop specific proteins to antagonize APOBECs -vif in HIV-1, for example. Evolving an entirely new protein function is much more difficult than, say, mutating an antibody binding site.
The comment from John Coffin you read on twievo 3 was quite thought-provoking! I felt ‘taken in’ as well. I hope you discuss viral mutation and recombination rates further.