
 If the reader does not believe that viroids and satellites are distinctive, then surely prions, infectious agents composed only of protein, must impress.
If the reader does not believe that viroids and satellites are distinctive, then surely prions, infectious agents composed only of protein, must impress.
The question of whether infectious agents exist without genomes arose with the discovery and characterization of infectious agents associated with a group of diseases called transmissible spongiform encephalopathies (TSEs). These diseases are rare, but always fatal, neurodegenerative disorders that afflict humans and other mammals. They are characterized by long incubation periods, spongiform changes in the brain associated with loss of neurons, and the absence of host responses. TSEs are caused by infectious proteins called prions.
The first TSE recognized was scrapie, so called because infected sheep tend to scrape their bodies on fences so much that they rub themselves raw. Scrapie has been recognized as a disease of European sheep for more than 250 years. It is endemic in some countries, for example, the United Kingdom, where it affects 0.5 to 1% of the sheep population each year.
Sheep farmers discovered that animals from affected herds could pass the disease to a scrapie-free herd, implicating an infectious agent. Infectivity from extracts of scrapie-affected sheep brains was shown to pass through filters with pores small enough to retain everything but viruses. As early as 1966, scrapie infectivity was shown to be considerably more resistant than that of most viruses to ultraviolet (UV) and ionizing radiation. Other TSE agents exhibit similar UV resistance. On the basis of this relative resistance to UV irradiation, some investigators argued that TSE agents are viruses well shielded from irradiation, whereas others claimed that TSE agents have little or no nucleic acids.
Several lines of evidence indicated that human spongiform encephalopathies might be caused by an infectious agent. Carleton Gajdusek and colleagues studied the disease kuru, found in the Fore people of New Guinea. This disease is characterized by cerebellar ataxia (defective motion or gait) without loss of cognitive functions. Kuru spread among women and children as a result of ritual cannibalism of the brains of deceased relatives. When cannibalism stopped in the late 1950s, kuru disappeared. Others observed that lesions in the brains of humans with kuru were similar to lesions in the brains of animals with scrapie. It was soon demonstrated that kuru and other human TSEs can be transmitted to chimpanzees and laboratory animals.
Human spongiform encephalopathies are placed into three groups: infectious, familial or genetic, and sporadic, distinguished by how the disease is acquired initially. An infectious (or transmissible) spongiform encephalopathy is exemplified by kuru and iatrogenic spread of disease to healthy individuals by transplantation of infected corneas, the use of purified hormones, or transfusion with blood from patients with the TSE Creutzfeldt-Jakob disease (CJD). Over 400 cases of iatrogenic Creutzfeldt-Jakob disease have been reported worldwide. The epidemic spread of bovine spongiform encephalopathy (mad cow disease, see below) among cattle in Britain can be ascribed to the practice of feeding processed animal by-products to cattle as a protein supplement. Similarly, the new human disease, variant CJD, arose after consumption of beef from diseased cattle. Sporadic CJD is a disease affecting one to five per million annually, usually late in life (with a peak at 68 years). As the name indicates, the disease appears with no warning or epidemiological indications. Kuru may have been originally established in the small population of Fore people in New Guinea when the brain of an individual with sporadic CJD was eaten. Familial spongiform encephalopathy is associated with an autosomal dominant mutation in the prnp gene. Together familial and sporadic forms of prion disease account for ~99% of all cases.
Clinical signs of infection commonly include cerebellar ataxia, memory loss, visual changes, dementia, and akinetic mutism, with death occurring after months or years. Once the infectious agent is in the central nervous system, the characteristic pathology includes severe astrocytosis, vacuolization (hence the term spongiform), and loss of neurons. There are no inflammatory, antibody, or cellular immune responses.
The unconventional physical attributes and slow infection pattern originally prompted many to argue that TSE agents are not viruses at all. In 1967 it was suggested that scrapie could be caused by a host protein, not by a nucleic acid-carrying virus.
An important breakthrough occurred in 1981, when characteristic fibrillar protein aggregates were visualized in infected brains. These aggregates could be concentrated by centrifugation and remained infectious. Stanley Prusiner and colleagues isolated a protein with unusual properties from scrapie-infected tissue. This protein is insoluble and relatively resistant to proteases. He named the scrapie infectious agent a prion, from the words protein and infectious.
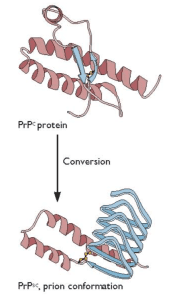
Prusiner’s unconventional proposal was that an altered form of a normal cellular protein, called PrPC, causes the fatal encephalopathy characteristic of scrapie. This controversial protein-only hypothesis caused a firestorm among those who study infectious disease. The hypothesis was that the essential pathogenic component is the host-encoded PrPC protein with an altered conformation, called PrPsc (€œPrP-scrapie€). Furthermore, in the simplest case, PrPSc was proposed to have the property of converting normal PrPC protein into more copies of the pathogenic form (illustrated). In recognition of his work on prions, Prusiner was awarded the Nobel Prize in physiology or medicine in 1997.
Sequence analysis of this protein led to the identification of the prnp gene, which is highly conserved in the genomes of many mammals, including humans. Expression of the prnp gene is now known to be essential for the pathogenesis of TSEs. The prnp gene encodes a 35-kDa membrane-associated neuronal glycoprotein, PrPC. The function of this protein has been difficult to determine because mice lacking both copies of the prnp gene develop normally and have few obvious defects. However, these mice are resistant to TSE infection, showing that PrPC is essential for prion propagation.
The discovery of the prnp gene has helped explain the basis of familial TSE diseases such as Creutzfeldt-Jakob disease, Gerstmann-Straussler-Scheinker disease, and fatal familial insomnia. Gerstmann-Straussler-Scheinker disease is associated with a change at PrPC amino acid 102 from proline to leucine. Introduction of this amino acid change into mice gives rise to a spontaneous neurodegenerative disease characteristic of a TSE. Familial Creutzfeldt-Jakob disease may be associated with an insertion of 144 base pairs at codon 53, or changes at amino acids 129, 178, or 200. In fatal familial insomnia, adults develop a progressive sleep disorder and typically die within one year. Development of this disease is strongly linked to the D178N amino acid change.
In the mid 1980s, a new disease appeared in cows in the United Kingdom: bovine spongiform encephalopathy, also called mad cow disease. It is believed to have been transmitted to cows by feeding them meat and bone meal, a high protein supplement prepared from the offal of sheep, cattle, pigs, and chicken. In the late 1970s the method of preparation of meat and bone meal was changed, resulting in material with a higher fat content. It is believed that this change allowed prions, from either a diseased sheep or cow, to retain infectivity and pass on to cattle. Before the disease was recognized in 1985, it was amplified by feeding cows the remains of infected bovine tissues: the incubation period for bovine spongiform encephalopathy is 5 years, but disease was not observed because most cattle are slaughtered between 2-3 years of age. Three years later, as the number of cases of mad cow disease increased, a ban on the use of meat and bone meal was put in place, a practice that together with culling infected cattle has stopped the epidemic. Over 180,000 cattle, mostly dairy cows, died of bovine spongiform encephalopathy from 1986-2000.
Cases of variant Creutzfeld-Jakob disease, a new TSE of humans, began to appear in 1994 in Great Britain. These were characterized by a lower mean age of the patients (26 years), longer duration of illness, and differences in other clinical and pathological characteristics. The results of epidemiologic and experimental studies indicate that variant Creutzfeld-Jakob disease is caused by prions transmitted by the consumption of cattle with bovine spongiform encephalopathy. As of 2011 there had been 175 cases of variant Creutzfeld-Jakob disease in the United Kingdom, and 215 globally.
Bovine spongiform encephalopathy continues to be detected in cattle. As of April 2012, 4 cases have been identified in the United States and 19 in Canada. These cases may arise sporadically, or through consumption of contaminated feed. Because cattle are slaughtered before disease symptoms are evident, there is concern that variant Creutzfeldt-Jakob might increase as contaminated meat enters the food supply. These concerns are being addressed by imposing bans on animal protein-containing feed, and increased surveillance of cows for the disease, for which diagnostic tests are being developed.
Chronic wasting disease is a transmissible spongiform encephalopathy of cervids such as deer, elk, and moose. It is the only known TSE to occur in free-ranging animals. The disease has been reported in the United States, Canada, and South Korea. In captive herds in the US and Canada up to 90% of mule deer and 60% of elk are infected, and the incidence in wild cervids is as high as 15%. Hunters are advised not to shoot or consume an elk or deer that is acting abnormally or appears to be sick, to avoid the brain and spinal cord when field dressing game, and not to consume brain, spinal cord, eyes, spleen, or lymph nodes. No case of transmission of chronic wasting disease prions to deer hunters has yet been reported.
It is not known how the disease is spread among cervids, but transmission by grass contaminated with saliva and feces is one possibility. When deer are fed prions they excrete them in the feces before developing clinical signs of infection, and prions can also be detected in deer saliva. In the laboratory, brain homogenates from infected deer can transmit the disease to cows. A concern is that prions of chronic wasting disease could be transmitted to cows grazing in pastures contaminated by cervids.
Since prions were discovered it has become clear that they cause a wider spectrum of neurodegenerative diseases. For example, the amyloid fibrils in Alzheimer’s disease contain the amyloid-beta peptide that is processed from the amyloid precursor protein; familial disease is caused by mutations in the gene for this protein. Mutations in the tau gene are responsible for heritable tauopathies including familial frontotemporal dementia and inherited progressive supranuclear palsy. Self-propagating tau aggregates pass from cell to cell. The prion-like spread of misfolded alpha-synuclein is believed to be involved in Parkinson’s disease. In these cases there is good evidence that the causative protein, like PrPSc, adopts a conformation that becomes self-propagating.
Despite the involvement of prions in human neurological diseases, in other organisms such proteins are not pathogenic but rather impart diverse functions through templated conformational change of a normal cellular protein. Such prions have been described in fungi where they do not form infectious particles and do not spread from cell to cell. These proteins change conformation in response to an environmental stimulus and acquire a new, beneficial function. An example is the Saccharomyces cerevisiae Ure2p protein, which normally is a nitrogen catabolite repressor when cells are grown in the presence of a rich source of nitrogen. In the aggregated prion state, called [URE3], the protein allows growth on poor nitrogen sources. These findings prompt the question of whether the conversion of PrPC to PrPSC once had a beneficial function that became pathogenic. If so, identifying that function, and how it was usurped, will be important for understanding the pathogenesis of transmissible spongiform encephalopathies.
Comments are closed.